
Bringing Machine Intelligence To Life
One main interest of c4Lab is to annotate variants and DNA sequences in the human genome. We built machine learning and deep learning models to predict variant pathogenicity, functional regions (e.g. enhancer, TFBS, eQTL, etc) and their sequence effect.
In c4Lab, we developed computational methods to solve biological problems, including GenEpi, ezGeno, VariantEpi, MHCfovea, and QuantEval.
We also built the variant database TaiwanGenomes and released TWB PRS models for studying Taiwanese geneatic data.

2025August 06Whole Exome Sequencing-identified Germline Variants Underlie High Familial Risk and Early-onset Colorectal Cancer in Taiwan
BackgroundGermline genetics contribute to colorectal cancer (CRC), influencing familial risk and early onset. However, such genetic contributions remain poorly characterized in Asian populations and have not been systematically studied in Taiwan.FindingsA total of 24 pathogenic variants in hereditary cancer genes were identified in 5.2% of Taiwanese patients with CRC, with prevalence rising to 8.2% in early-onset cases. Additionally, 25.5% of rare-risk variants were novel, including 41 candidate pathogenic variants linked to familial or early-onset CRC.Implications for patient careThis study revealed the germline genetic landscape of CRC in Taiwan, identifying novel and population-specific variants that enhance hereditary risk assessment. These findings support more tailored screening strategies to improve early detection and outcomes in Asian populations.
2025Feb 10深度學習應用於多體學分析加速個人基因體註解|基因體醫學月會
The analysis of personal multi-omics data is pivotal in advancing precision medicine. AI-powered multi-omics data analysis accelerates our understanding of human gene functions and the impacts of personal genetic variants on these functional elements. This talk will begin with an overview of the data flow from genome, epigenome, and transcriptome to proteome. Following this, I will discuss the types of high-throughput multi-omics data generated by next-generation sequencing since 2006. Recent advances in deep learning methods have shown great promise in addressing challenging problems in computational biology, such as protein structure prediction and gene regulation modeling. These developments in deep neural networks are poised to significantly enhance clinical practices in precision medicine in the near future.
2025Feb 03Annotation-free deep learning for predicting gene mutations from whole slide images of acute myeloid leukemia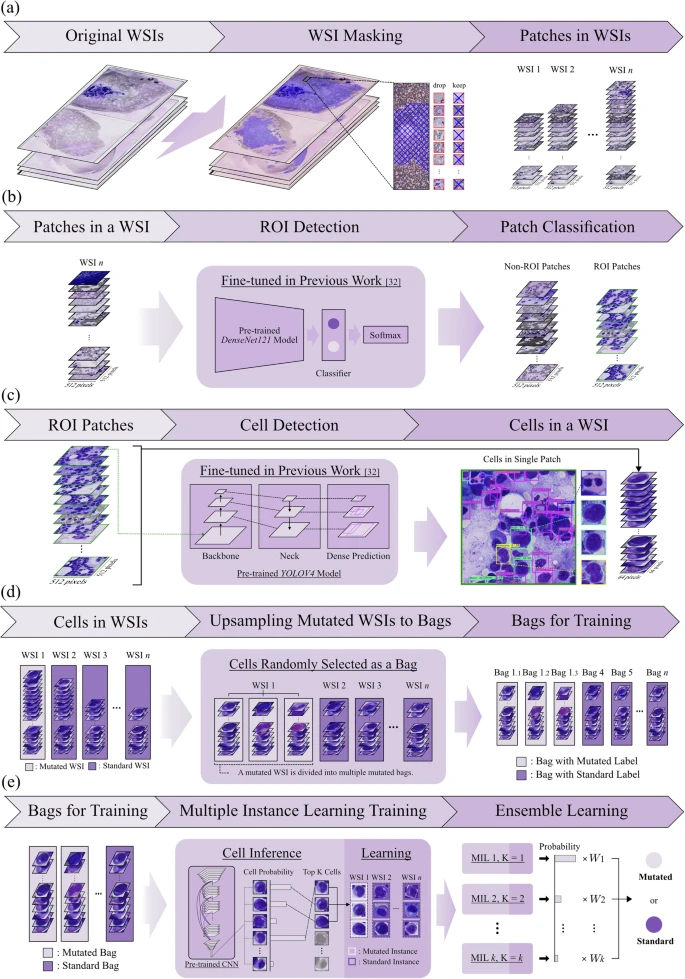
The rapid development of deep learning has revolutionized medical image processing, including analyzing whole slide images (WSIs). Despite the demonstrated potential for characterizing gene mutations directly from WSIs in certain cancers, challenges remain due to image resolution and reliance on manual annotations for acute myeloid leukemia (AML). We, therefore, propose a deep learning model based on multiple instance learning (MIL) with ensemble techniques to predict gene mutations from AML WSIs. Our model predicts NPM1 mutations and FLT3-ITD without requiring patch-level or cell-level annotations. Using a dataset of 572 WSIs, the largest database with both WSI and genetic mutation information, our model achieved an AUC of 0.90 ± 0.08 for NPM1 and 0.80 ± 0.10 for FLT3-ITD in the testing cohort. Additionally, we found that blasts are pivotal indicators for gene mutation predictions, with their proportions varying between mutated and standard WSIs, highlighting the clinical potential of AML WSI analysis.
2024Aug 01【人工智慧驅動的多體學分析於精準醫學之應用
AI-Driven Multi-Omics Analysis in Precision Medicine 】Chien-Yu Chen
The analysis of personal multi-omics data is pivotal in advancing precision medicine. AIpowered multi-omics data analysis accelerates our understanding of human gene functions and the impacts of personal genetic variants on these functional elements. This talk will begin with an overview of the data flow from genome, epigenome, and transcriptome to proteome. Following this, I will discuss the types of high-throughput multi-omics data generated by next-generation sequencing since 2006. Recent advances in deep learning methods have shown great promise in addressing challenging problems in computational biology, such as protein structure prediction and gene regulation modeling. These developments in deep neural networks are poised to significantly enhance clinical practices in precision medicine in the near future.
2023Dec 29Complete genomic profiles of 1496 Taiwanese reveal curated medical insights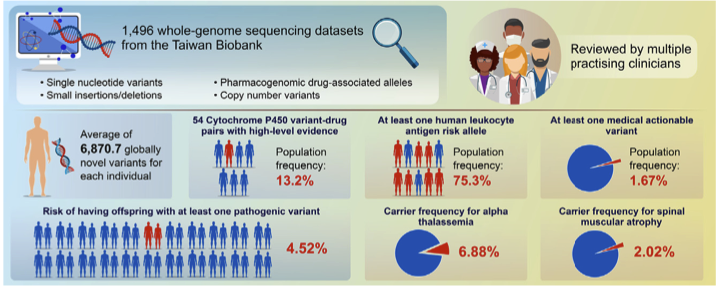
We found that each participant had an average of 6,870.7 globally novel variants and 75.3% (831/1103) of the participants harbored at least one PharmGKB-selected high evidence level human leukocyte antigen (HLA) risk allele. 54 PharmGKB-reported high-level instances of evidence of Cytochrome P450 variant-drug pairs, with a population frequency of over 13.2%. We also identified 23 variants in the ACMG secondary finding V3 gene list from 25 participants, suggesting that 1.67% (25/1496) of the population is harboring at least one medical actionable variant. Our carrier status analyses suggest that one in 25 couples (3.94%) would risk having offspring with at least one pathogenic variant, which is in line with rates found in Japan and Singapore. For pathogenic CNV, we detected 6.88% and 2.02% carrier rates for alpha thalassemia and spinal muscular atrophy, respectively.
2022May 19AI for Life Science and Precision Medicine
Outline: AI to predict molecular binding
*MHCfovea (https://mhcfovea.ailabs.tw/)
*ezGeno (https://github.com/ailabstw/ezGeno)
AI to read biomedical literature
*pubmedKB (https://www.pubmedkb.cc/)
AI to estimate disease risks
*TaiwanGenomes (https://genomes.tw/)
*TWB-PRS (https://github.com/chienyuchen/TWB-PRS/)
2021Aug 16An AutoML solution for Epigenomics analysis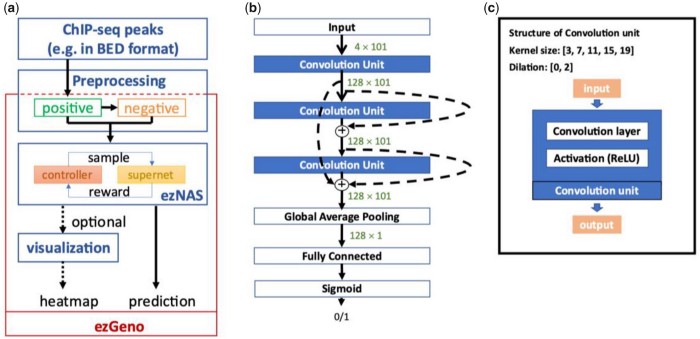
To facilitate the process of tailor-making a deep neural network for exploring the dynamics of genomic DNA, we have developed a hands-on package called ezGeno. ezGeno automates the search process of various parameters and network structures and can be applied to any kind of 1D genomic data. Combinations of multiple abovementioned 1D features are also applicable. The ezGeno package can be freely accessed at https://github.com/ailabstw/ezGeno.
2020Dec 09人工智慧在生物資訊的應用︱大安高工
本演講首先介紹開發 AlphaGo 的 DeepMind 團隊如何運用類似的人工智慧技術 預測蛋白質 3D結構,這是一個非常重要且歷史悠久的生物資訊計算問題,這兩年因為這波人工智慧的浪潮推動而有了突破性的發展。此演講中也將分享,個人DNA中的突變 (mutation) 是否可能影響蛋白質結構,進而影響蛋白質的功能,不正確的 3D 結構將造成疾病,而這些突變所產生的影響都可以使用人工智慧演算法預測評估。不僅如此,癌細胞中的突變,也是醫師進行用藥選擇的重要依據,我們期待未來有越來越多的基因數據累積,人工智慧將有更大的發揮空間,幫助疾病的預防或選擇最合適的治療方案。
2020Oct 09DockCoV2: a drug database against SARS-CoV-2
We built DockCoV2 to provide the binding affinity of FDA-approved and Taiwan National Health Insurance (NHI) drugs with seven proteins. This database contains a total of 3,109 drugs. DockCoV2 is available at https://covirus.cc/drugs/
2020Feb 24GenEpi: gene-based epistasis discovery using machine learning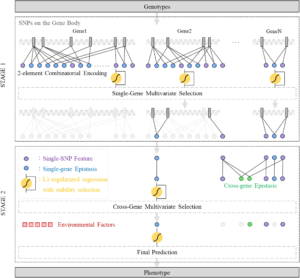
We developed an efficient and effective GWAS method to detect epistasis for discovering sophisticated pathogenesis, which is especially important for complex diseases such as Alzheimer’s disease (AD). GenEpi is a computational package to uncover epistasis associated with phenotypes by the proposed machine learning approach. GenEpi identifies both within-gene and cross-gene epistasis through a two-stage modeling workflow. In both stages, GenEpi adopts two-element combinatorial encoding when producing features and constructs the prediction models by L1-regularized regression with stability selection. The simulated data showed that GenEpi outperforms other widely-used methods on detecting the ground-truth epistasis. As real data is concerned, this study uses AD as an example to reveal the capability of GenEpi in finding disease-related variants and variant interactions that show both biological meanings and predictive power.
2019Jun 05Effect of de novo transcriptome assembly on transcript quantification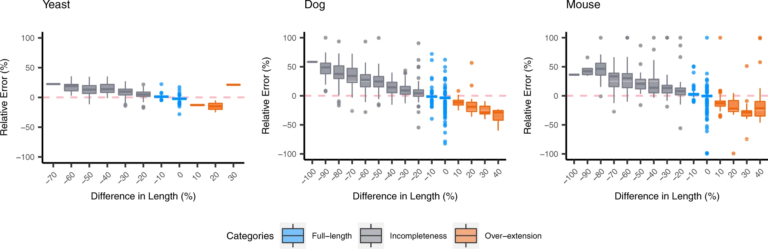
For the organisms without the reference transcriptome, de novo transcriptome assembly must be carried out prior to quantification. This study investigates how assembly quality affects the performance of quantification based on de novo transcriptome assembly. We examined the over-extended and incomplete contigs, and demonstrated that assembly completeness has a strong impact on the estimation of contig abundance. Then we investigated the behavior of the quantifiers with respect to sequence ambiguity which might be originally presented in the transcriptome or accidentally produced by assemblers. The results suggested that the quantifiers often over-estimate the expression of family-collapse contigs and under-estimate the expression of duplicated contigs. For organisms without reference transcriptome, it remains challenging to detect the inaccurate estimation on family-collapse contigs. On the contrary, we observed that the situation of under-estimation on duplicated contigs can be warned through analyzing the read proportion of estimated abundance (RPEA) of contigs in the connected component inferenced by the quantifiers. In addition, we suggest that the estimated quantification results on the connected component level have better accuracy over sequence level quantification.
2019Jan 23用人工智慧探索DNA中的調控密碼 / 陳倩瑜教授
DNA是所有生命進行細胞活動的藍圖,和我們所熟悉的二進位數位系統類似,生命利用ACGT四個字母編碼建構四進位系統,引導細胞內的分子進行複雜的交互作用與化學反應。不同於人類製造的二進位數位系統,生命的四進位系統是大自然長期演化的成品,DNA中有非常多重要的功能模組是大自然精心設計的傑作,近幾年拜許多新穎的高通量生物技術之賜,科學家得以一窺這些藍圖的全貌,並監測細胞中各式各樣的生命狀態,這些生醫大數據快速累積,於是有了機器學習與人工智慧可以揮灑的空間。本演講將以調控密碼為例,粗淺地與大家分享人工智慧在生物資訊領域的應用。
2018May 02Whole-genome de novo sequencing reveals unique genes that contributed to the adaptive evolution of the Mikado pheasant
攝影:謝郁震
The Mikado pheasant 帝雉 (Syrmaticus mikado) is a nearly endangered species indigenous to high-altitude regions of Taiwan. We completed the draft genome of the Mikado pheasant, which consists of 1.04 Gb of DNA and 15,972 annotated protein-coding genes. The Mikado pheasant displays expansion and positive selection of genes related to features that contribute to its adaptive evolution, such as energy metabolism, oxygen transport, hemoglobin binding, radiation response, immune response, and DNA repair. To investigate the molecular evolution of the major histocompatibility complex (MHC) across several avian species, 39 putative genes spanning 227 kb on a contiguous region were annotated and manually curated. The MHC loci of the pheasant revealed a high level of synteny, several rapidly evolving genes, and inverse regions compared to the same loci in the chicken. The complete mitochondrial genome was also sequenced, assembled, and compared against four long-tailed pheasants. The results from molecular clock analysis suggest that ancestors of the Mikado pheasant migrated from the north to Taiwan about 3.47 million years ago.
2017Dec 08[Dosudo] DeepLearning MEET #11 陳倩瑜教授專訪
隨著基因定序技術的成熟及便利, 基因組資料已經變成深度學習下一個最受矚目及最具潛力的焦點, 各大公司也紛紛佈局在AI醫療及基因研究上. 這次 Dosudo 的專訪榮幸的邀請到臺大生物產業機電工程學系的陳倩瑜教授來為我們談談如何將深度學習技術應用在基因資料的分析, 以及為何生物資訊需要結合人工智慧及這領域未來的展望.
2017May 23當深度學習與生物資訊在個人全基因體註解相遇
Bioinformatics has played an important role in annotating the human genome since its draft was first announced in 2001. As the sequencing cost decreased dramatically owing to the advance of next-generation sequencing technology, the need of precisely annotating a personal genome is right around the corner. This talk will start with the success of using structural bioinformatics in predicting the influence of a single nucleotide variation on changing the protein-DNA binding affinity. Next, the concept of deep learning and how it has been used to annotate epigenomes and to explore the roles of cis-regulatory sequence variations will be introduced. As the scale and complexity of personal genomic data analysis increase rapidly, deep learning will definitely become one of the effective ways to associate personal genomic variations with diseases or drug responses. The current status and challenges of using deep learning in annotating personal genomes will be kindly addressed in the talk and it deserves more attentions when designing Bioinformatics education in the near future.
2016Integrating RNA-seq and ChIP-seq Data to Characterize Long Non-coding RNAs in Drosophila melanogaster
Recent advances in sequencing technology have opened a new era in RNA studies. Novel types of RNAs such as long non-coding RNAs (lncRNAs) have been discovered by transcriptomic sequencing and some lncRNAs have been found to play essential roles in biological processes. However, only limited information is available for lncRNAs in Drosophila melanogaster, an important model organism. Therefore, the characterization of lncRNAs and identification of new lncRNAs in D. melanogaster is an important area of research. Moreover, there is an increasing interest in the use of ChIP-seq data (H3K4me3, H3K36me3 and Pol II) to detect signatures of active transcription for reported lncRNAs.
In this study, we have developed a computational approach to identify new lncRNAs from two tissue-specific RNA-seq datasets using the poly(A)-enriched and the ribo-zero method, respectively. In our results, we identified 462 novel lncRNA transcripts, which we combined with 4137 previously published lncRNA transcripts into a curated dataset. We then utilized 61 RNA-seq and 32 ChIP-seq datasets to improve the annotation of the curated lncRNAs with regards to transcriptional direction, exon regions, classification, expression in the brain, possession of a poly(A) tail, and presence of conventional chromatin signatures. Furthermore, we used 30 time-course RNA-seq datasets and 32 ChIP-seq datasets to investigate whether the lncRNAs reported by RNA-seq have active transcription signatures. The results showed that more than half of the reported lncRNAs did not have chromatin signatures related to active transcription. To clarify this issue, we conducted RT-qPCR experiments and found that ~95.24 % of the selected lncRNAs were truly transcribed, regardless of whether they were associated with active chromatin signatures or not.
2015Discovery of Genes Related to Formothion Resistance in Oriental Fruit Fly (Bactrocera dorsalis) by a Constrained Functional Genomics Analysis
Oriental fruit fly is a very destructive pest of fruit in areas where it occurs and some of them have insecticide resistance.
The underlying basis of such phenomena can involve complex interactions of multiple genes. This study aims to analyze the specific genes are involved in resistance mechanisms.
2013De novo transcriptome sequencing of axolotl blastema for identification of differentially expressed genes during limb regeneration
It’s amazing for salamanders are unique among vertebrates in their ability to completely regenerate amputated limbs through the mediation of blastema cells located at the stump ends. This study aims to know the secret in transcription mechanism of salamander’s regeneration.
2013PiDNA: predicting protein-DNA interactions with structural models
Predicting binding sites of a transcription factor in the genome is an important, but challenging, issue in studying gene regulation. PiDNA, aims at first constructing reliable PWMs by applying an atomic-level knowledge-based scoring function on numerous in silico mutated complex structures, and then using the PWM constructed by the structure models with small energy changes to predict the interaction between proteins and DNA sequences. This study first shows that the constructed PWMs are similar to the annotated PWMs collected from databases or literature. Second, the prediction accuracy of PiDNA in detecting relatively high-specificity sites is evaluated by comparing the ranked lists against in vitro experiments from protein-binding microarrays. Finally, PiDNA is shown to be able to select the experimentally validated binding sites from 10 000 random sites with high accuracy.
PiDNA is available at:
here.
2012機器學習的演進與應用 / 陳倩瑜教授
機器能跟人類一樣有智慧嗎?近年來,隨著數位化資料的大量累積,人工智慧(AI)的子領域機器學習(machine learning)發展迅速,機器已經能自己從大量資料中學習,「看起來」比以前更有智慧。怎麼讓機器自己學習呢?專長生物資訊的陳教授將為我們介紹機器學習的定義與演進,並帶我們探索機器學習的多元應用,瞭解除了搜尋引擎與智慧型手機之外,機器學習如何輔助基礎醫學研究:探索基因與各種疾病之關聯性,將在未來個人化醫療時代中扮演關鍵角色。
2012Discovery of genes related to insecticide resistance in Bactrocera dorsalis by functional genomic analysis of a de novo assembled transcriptome
Insecticide resistance has recently become a critical concern for control of many insect pest species. Genome sequencing and global quantization of gene expression through analysis of the transcriptome can provide useful information relevant to this challenging problem. This study aims at using whole transcriptome analysis developed through de novo assembly to know resistance mechanisms from molecular aspect.
2012De novo motif discovery facilitates identification of interactions between transcription factors in Saccharomyces cerevisiae
Previous studies have used target genes shared by two TFs as a clue to infer TF-TF interactions. However, the target genes with low binding affinity are frequently omitted by experimental data, especially when a single strict threshold is employed. This study aims at improving the accuracy of inferring TF-TF interactions.
2012Predicting Target DNA Sequences of DNA-binding Proteins Based on Unbound Structures
DNA-binding proteins such as transcription factors use DNA-binding domains (DBDs) to bind to specific sequences in the genome to initiate many important biological functions. This study aims at investigating the possibility of predicting the DNA sequences bound by DNA-binding proteins from the proteins’ unbound structures (structures of the unbound state).
2011WildSpan: Mining Structured Motifs from Protein Sequences
Automatic extraction of motifs from biological sequences is an important research problem in study of molecular biology. For proteins, it is desired to discover sequence motifs containing a large number of wildcard symbols, as the residues associated with functional sites are usually largely separated in sequences. This paper proposes an algorithm named WildSpan (sequential pattern mining across large wildcard regions) that incorporates several pruning strategies to largely reduce the mining cost.
2008 Discovering gapped binding sites of yeast transcription factors
We propose a method for discovering TFBSs, especially gapped motifs. We use ChIP-chip data to judge the binding strength of a TF to a putative target promoter and use orthologous sequences from related species to judge the degree of evolutionary conservation of a predicted TFBS.
2006Protein Disorder Prediction by Condensed PSSM Considering Propensity for Order or Disorder
More and more disordered regions have been discovered in protein sequences, and many of them are found to be functionally significant. Previous studies reveal that disordered regions of a protein can be predicted by its primary structure, the amino acid sequence.
Recent studies further show that employing evolutionary information such as position specific scoring matrices (PSSMs) improves the prediction accuracy of protein disorder. As more and more machine learning techniques have been introduced to protein disorder detection, extracting more useful features with biological insights attracts more attention.
2005c4Lab was founded
c4Lab was founded in the department of Biomechartronics Engineering, National Taiwan University by Dr. Chien-Yu Chen and her students.
2004Incremental Generation of Summarized Clustering Hierarchy for Protein Family Analysis
Protein sequence clustering has been widely exploited to facilitate in-depth analysis of protein functions and families. For some applications of protein sequence clustering, it is highly desirable that a hierarchical structure, also referred to as dendrogram, which shows how proteins are clustered at various levels, is generated. In this paper, the design of a novel incremental clustering algorithm aimed at generating summarized dendrograms for analysis of protein databases is described. The proposed incremental clustering algorithm employs a statistics-based model to summarize the distributions of the similarity scores among the proteins in the database and to control formation of clusters.
CONTACT US
TEL
+886-2-3366-7118
(Prof. Chen, Chien-Yu)
ADD
R304 Department of Biomechatronics
(new building), National Taiwan University
Onlayn mühitdə Mostbet mükəmməl bahis rahat və sürətli istifadə üçün nəzərdə tutulub.