From 2015
Sequence/Variant Annotation
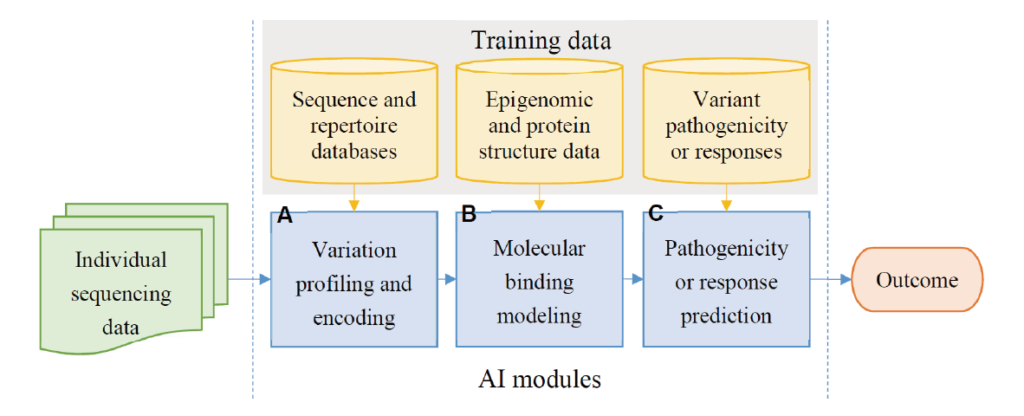
One main interest of our lab is to annotate variants and DNA sequences from human genome. We’ve built machine learning and deep learning models to predict variant pathogenicity, functional regions (e.g. enhancer, TFBS, eQTL, etc) and their sequence effect.
RESEARCH CONTENT
- A number of databases contain a growing number of disease variants that are continuously being discovered in the human genome, such as Clinvar, OMIM and HGMD. ACMG offered an evidence framework and criteria for classifying pathogenic variants. As validation with clinical laboratories may take much effort, the annotation for disease variants pathogenicity has been studied with a variety of in silico tools based on the concept of the evolutionary conservation and protein function change of the variant effect. However, the imbalanced data for positive and negative biological samples remains a problem in the classification task. To correctly identify wether a non-coding variant is pathogenic or not, resampling techniques considering the feature space and cluster ensemble methods showed a promising result from our previous work CE-SMURF.
- One class of the regulatory elements that have been shown to act as key components to assist promoters in modulating the gene expression in living cells are enhancers. Enhancers function by looping to the promoter that they regulate, and are activated by sequence-specific transcription factors, which create an environment permissive to transcription. At present, the enhancer regions are identified from the chromatin structures, which can be analyzed by Chromosome conformation capture techniques or infered from histone modification signals and transcription factor binding sites. To leverage the extensive experimental data to the functional region, we developed a deep learning model, accuEnhancer, to learn the enhancer activity of H3k27ac signals from genomic sequences and the DNase signals of each corresponding region. Furthermore, we extended the model from within-cell type to cross-cell type to perform superior to the existing enhancer prediction tools.
- Functional annotation of specific regions of the genomic sequences are evaluated from a variety of empigenomic signals in order to predict chromatin activities, TF binding or expression states. After successfully training models from the accumulated large amount of experimental datasets of expression and histone modification profiles for quantification prediction, mutating variant positions inside target sequences may change the predicting profiles. A significant difference between the reference and alternative sequence profile predictions suggest a corresponding functional gain or loss from the variant effect. Constructing this process to a pipeline helps biologists to better evaluate the disease/phenotype related variants and find cure or treatment to it.